In almost all Fe–S proteins, the Fe centers are tetrahedral and the terminal ligands are thiolato sulfur centers from cysteinyl residues. The sulfide groups are either two- or three-coordinated. Three distinct kinds of Fe–S clusters with these features are most common.
Iron-sulfur proteins are involved in various biological electron transport processes, such as photosynthesis and cellular respiration, which require rapid electron transfer to sustain the energy or biochemical needs of the organism. To serve their various biological roles, iron-sulfur proteins effect rapid electron transfers and span the whole range of physiological redox potentials from -600 mV to +460 mV.
Fe3+-SR bonds have unusually high covalency which is expected.[according to whom?] When comparing the covalency of Fe3+ with the covalency of Fe2+, Fe3+ has almost double the covalency of Fe2+ (20% to 38.4%).[5] Fe3+ is also much more stabilized than Fe2+. Hard ions like Fe3+ normally have low covalency because of the energy mismatch of the metal lowest unoccupied molecular orbital with the ligand highest occupied molecular orbital.
External water molecules positioned close to the iron-sulfur active site reduces covalency; this can be shown by lyophilization experiments where water is removed from the protein. This reduction is because external water hydrogen bonds with cysteine S, decreasing the latter's lone pair electron donation to the Fe3+/2+ by pulling away S electrons.[5] Since covalency stabilizes Fe3+ more than Fe2+, Fe3+ is more destabilized by the HOH-S hydrogen-bonding.
The Fe3+ 3d orbital energies follow the "inverted" bonding scheme which fortuitously has the Fe3+ d-orbitals closely matched in energy with the sulfur 3p orbitals, giving high covalency in the resulting bonding molecular orbital.[3] This high covalency lowers the inner sphere reorganization energy[3] and ultimately contributes to a rapid electron transfer.
The simplest polymetallic system, the [Fe2S2] cluster, is constituted by two iron ions bridged by two sulfide ions and coordinated by four cysteinylligands (in Fe2S2ferredoxins) or by two cysteines and two histidines (in Rieske proteins). The oxidized proteins contain two Fe3+ ions, whereas the reduced proteins contain one Fe3+ and one Fe2+ ion. These species exist in two oxidation states, (FeIII)2 and FeIIIFeII. CDGSH iron sulfur domain is also associated with 2Fe-2S clusters.
Rieske 2Fe-2S Cluster Oxidation States of Fe3+ and Fe2+
The Rieske proteins contain Fe–S clusters that coordinate as a 2Fe–2S structure and can be found in the membrane bound cytochrome bc1 complex III in the mitochondria of eukaryotes and bacteria. They are also a part of the proteins of the chloroplast such as the cytochrome b6f complex in photosynthetic organisms. These photosynthetic organisms include plants, green algae, and cyanobacteria, the bacterial precursor to chloroplasts. Both are part of the electron transport chain of their respective organisms which is a crucial step in the energy harvesting for many organisms.[6]
A common motif features a four iron ions and four sulfide ions placed at the vertices of a cubane-type cluster. The Fe centers are typically further coordinated by cysteinyl ligands. The [Fe4S4] electron-transfer proteins ([Fe4S4] ferredoxins) may be further subdivided into low-potential (bacterial-type) and high-potential (HiPIP) ferredoxins. Low- and high-potential ferredoxins are related by the following redox scheme:
4Fe-4S clusters serve as electron-relays in proteins.
In HiPIP, the cluster shuttles between [2Fe3+, 2Fe2+] (Fe4S42+) and [3Fe3+, Fe2+] (Fe4S43+). The potentials for this redox couple range from 0.4 to 0.1 V. In the bacterial ferredoxins, the pair of oxidation states are [Fe3+, 3Fe2+] (Fe4S4+) and [2Fe3+, 2Fe2+] (Fe4S42+). The potentials for this redox couple range from −0.3 to −0.7 V. The two families of 4Fe–4S clusters share the Fe4S42+ oxidation state. The difference in the redox couples is attributed to the degree of hydrogen bonding, which strongly modifies the basicity of the cysteinyl thiolate ligands.[citation needed] A further redox couple, which is still more reducing than the bacterial ferredoxins is implicated in the nitrogenase.
Some 4Fe–4S clusters bind substrates and are thus classified as enzyme cofactors. In aconitase, the Fe–S cluster binds aconitate at the one Fe centre that lacks a thiolate ligand. The cluster does not undergo redox, but serves as a Lewis acid catalyst to convert citrate to isocitrate. In radical SAM enzymes, the cluster binds and reduces S-adenosylmethionine to generate a radical, which is involved in many biosyntheses.[7]
4Fe-4S Oxidation States of Fe3+, Fe2.5+, and Fe2+.
The second cubane shown here with mixed valence pairs (2 Fe3+ and 2 Fe2+), has a greater stability from covalent communication and strong covalent delocalization of the “extra” electron from the reduced Fe2+ that results in full ferromagnetic coupling.
Proteins are also known to contain [Fe3S4] centres, which feature one iron less than the more common [Fe4S4] cores. Three sulfide ions bridge two iron ions each, while the fourth sulfide bridges three iron ions. Their formal oxidation states may vary from [Fe3S4]+ (all-Fe3+ form) to [Fe3S4]2− (all-Fe2+ form). In a number of iron–sulfur proteins, the [Fe4S4] cluster can be reversibly converted by oxidation and loss of one iron ion to a [Fe3S4] cluster. E.g., the inactive form of aconitase possesses an [Fe3S4] and is activated by addition of Fe2+ and reductant.
Examples include the active sites of a number of enzymes:
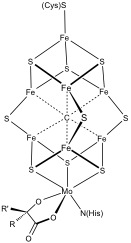
Structure of the FeMoco cluster in nitrogenase. The cluster is linked to the protein by the amino acid residues cysteine and histidine.Nitrogenase include two P-clusters ([8Fe-7S]) and two FeMocos ([7Fe-9S-C-Mo-R homocitrate]).[8]
[FeFe]-hydrogenase features an "H-cluster", consisting of a Fe4S4 bridge to Fe2 via a cystine. The Fe2 half features unique ligands: 3 CO, 2 CN-, and an azadithiolate HN(CH2S−)2.[11]
A special 6 cysteine-coordinated [Fe4S3] cluster was found in oxygen-tolerant membrane-bound [NiFe] hydrogenases.[12][13]
The "double cubane cluster" [Fe8S9], found in some nitrogenase-related ATPases, consists of two [Fe4S4] bridged by a cysteine. The functions of such proteins remain unclear.[14]
Ranges of reduction potentials, Eo (mV), covered by the different classes of iron-sulfur proteins, heme proteins, and copper proteins. (HiPIP = High potential iron-sulfur proteins, Rdx = rubredoxins, Fdx = ferredoxins, Cyt = cytochromes.)
The biosynthesis of the Fe–S clusters has been well studied.[15][16][17]
The biogenesis of iron sulfur clusters has been studied most extensively in the bacteria E. coli and A. vinelandii and yeast S. cerevisiae. At least three different biosynthetic systems have been identified so far, namely nif, suf, and isc systems, which were first identified in bacteria. The nif system is responsible for the clusters in the enzyme nitrogenase. The suf and isc systems are more general.
The yeast isc system is the best described. Several proteins constitute the biosynthetic machinery via the isc pathway. The process occurs in two major steps:
(1) the Fe/S cluster is assembled on a scaffold protein followed by (2) transfer of the preformed cluster to the recipient proteins.
The first step of this process occurs in the cytoplasm of prokaryotic organisms or in the mitochondria of eukaryotic organisms. In the higher organisms the clusters are therefore transported out of the mitochondrion to be incorporated into the extramitochondrial enzymes. These organisms also possess a set of proteins involved in the Fe/S clusters transport and incorporation processes that are not homologous to proteins found in prokaryotic systems.
Synthetic analogues of the naturally occurring Fe–S clusters were first reported by Holm and coworkers.[18] Treatment of iron salts with a mixture of thiolates and sulfide affords derivatives such as (Et4N)2Fe4S4(SCH2Ph)4].[19][20]
^Guan, Y.; Manuel, R. C.; Arvai, A. S.; Parikh, S. S.; Mol, C. D.; Miller, J. H.; Lloyd, S.; Tainer, J. A. (December 1998). "MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily". Nature Structural Biology. 5 (12): 1058–1064. doi:10.1038/4168. ISSN1072-8368. PMID9846876. S2CID22085836.
^Susan C. Wang; Perry A. Frey (2007). "S-adenosylmethionine as an oxidant: the radical SAM superfamily". Trends in Biochemical Sciences. 32 (3): 101–10. doi:10.1016/j.tibs.2007.01.002. PMID17291766.