Restriction site associated DNA (RAD) markers are a type of genetic marker which are useful for association mapping, QTL-mapping, population genetics, ecological genetics and evolutionary genetics. The use of RAD markers for genetic mapping is often called RAD mapping. An important aspect of RAD markers and mapping is the process of isolating RAD tags, which are the DNA sequences that immediately flank each instance of a particular restriction site of a restriction enzyme throughout the genome.[1] Once RAD tags have been isolated, they can be used to identify and genotype DNA sequence polymorphisms mainly in form of single nucleotide polymorphisms (SNPs).[1] Polymorphisms that are identified and genotyped by isolating and analyzing RAD tags are referred to as RAD markers. Although genotyping by sequencing presents an approach similar to the RAD-seq method, they differ in some substantial ways.[2][3][4]
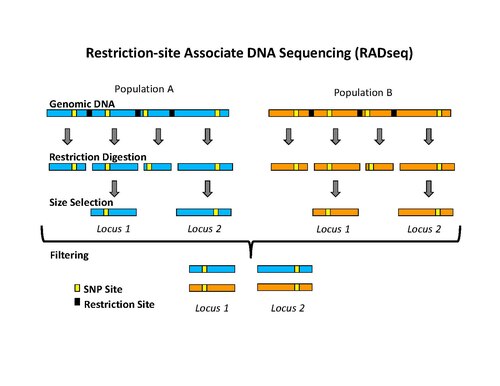
Isolation of RAD tags
editThe use of the flanking DNA sequences around each restriction site is an important aspect of RAD tags.[1] The density of RAD tags in a genome depends on the restriction enzyme used during the isolation process.[5] There are other restriction site marker techniques, like RFLP or amplified fragment length polymorphism (AFLP), which use fragment length polymorphism caused by different restriction sites, for the distinction of genetic polymorphism. The use of the flanking DNA-sequences in RAD tag techniques is referred as reduced-representation method.[2]
The initial procedure to isolate RAD tags involved digesting DNA with a particular restriction enzyme, ligating biotinylated adapters to the overhangs, randomly shearing the DNA into fragments much smaller than the average distance between restriction sites, and isolating the biotinylated fragments using streptavidin beads.[1] This procedure was used initially to isolate RAD tags for microarray analysis.[1][6][7] More recently, the RAD tag isolation procedure has been modified for use with high-throughput sequencing on the Illumina platform, which has the benefit of greatly reduced raw error rates and high throughput.[5] The new procedure involves digesting DNA with a particular restriction enzyme (for example: SbfI, NsiI,…), ligating the first adapter, called P1, to the overhangs, randomly shearing the DNA into fragments much smaller than the average distance between restriction sites, preparing the sheared ends into blunt ends and ligating the second adapter (P2), and using PCR to specifically amplify fragments that contain both adapters. Importantly, the first adapter contains a short DNA sequence barcode, called MID (molecular identifier) that is used as a marker to identify different DNA samples that are pooled together and sequenced in the same reaction.[5][8] The use of high-throughput sequencing to analyze RAD tags can be classified as reduced-representation sequencing, which includes, among other things, RADSeq (RAD-Sequencing).[2]
Detection and genotyping of RAD markers
editOnce RAD tags have been isolated, they can be used to identify and genotype DNA sequence polymorphisms such as single nucleotide polymorphisms (SNPs).[1][5] These polymorphic sites are referred to as RAD markers. The most efficient way to find RAD tags is by high-throughput DNA sequencing,[5][8] called RAD tag sequencing, RAD sequencing, RAD-Seq, or RADSeq.
Prior to the development of high-throughput sequencing technologies, RAD markers were identified by hybridizing RAD tags to microarrays.[1][6][7] Due to the low sensitivity of microarrays, this approach can only detect either DNA sequence polymorphisms that disrupt restriction sites and lead to the absence of RAD tags or substantial DNA sequence polymorphisms that disrupt RAD tag hybridization. Therefore, the genetic marker density that can be achieved with microarrays is much lower than what is possible with high-throughput DNA-sequencing.[9]
History
editRAD markers were first implemented using microarrays and later adapted for NGS (Next-Generation-Sequencing).[9] It was developed jointly by Eric Johnson and William Cresko's laboratories at the University of Oregon around 2006. They confirmed the utility of RAD markers by identifying recombination breakpoints in D. melanogaster and by detecting QTLs in threespine sticklebacks.[1]
ddRADseq
editIn 2012 a modified RAD tagging method called double digest RADseq (ddRADseq) was suggested.[10][11] By adding a second restriction enzyme, replacing the random shearing, and a tight DNA size selection step it is possible to perform low-cost population genotyping. This can be an especially powerful tool for whole-genome scans for selection and population differentiation or population adaptation.[11]
hyRAD
editA study in 2016 presented a novel method called hybridization RAD (hyRAD),[12] where biotinylated RAD fragments, covering a random fraction of the genome, are used as baits for capturing homologous fragments from genomic shotgun sequencing libraries. DNA fragments are first generated using ddRADseq protocol applied to fresh samples, and used as hybridization-capture probes to enrich shotgun libraries in the fragments of interest. This simple and cost-effective approach allows sequencing of orthologous loci even from highly degraded DNA samples, opening new avenues of research in the field of museomics. Another advantage of the method is not relying on the restriction site presence, improving among-sample loci coverage. The technique was first tested on museum and fresh samples of Oedaleus decorus, a Palearctic grasshopper species, and later implemented in regent honeyeater,[13] arthropods,[14] among other species. A lab protocol was developed to implement hyRAD in birds.[15]
See also
editReferences
edit- ^ a b c d e f g h Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA (February 2007). "Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers". Genome Research. 17 (2): 240–248. doi:10.1101/gr.5681207. PMC 1781356. PMID 17189378.
- ^ a b c Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (June 2011). "Genome-wide genetic marker discovery and genotyping using next-generation sequencing". Nature Reviews. Genetics. 12 (7): 499–510. doi:10.1038/nrg3012. PMID 21681211. S2CID 15080731.
- ^ Campbell EO, Brunet BM, Dupuis JR, Sperling FA (2018). "Would an RRS by any other name sound as RAD?". Methods in Ecology and Evolution. 9 (9): 1920–1927. doi:10.1111/2041-210X.13038.
- ^ Vaux F, Dutoit L, Fraser CI, Waters JM (2022). "Genotyping-by-sequencing for biogeography". Journal of Biogeography. 50 (2): 262–281. doi:10.1111/jbi.14516.
- ^ a b c d e Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. (2008). "Rapid SNP discovery and genetic mapping using sequenced RAD markers". PLOS ONE. 3 (10): e3376. Bibcode:2008PLoSO...3.3376B. doi:10.1371/journal.pone.0003376. PMC 2557064. PMID 18852878.
- ^ a b Miller MR, Atwood TS, Eames BF, Eberhart JK, Yan YL, Postlethwait JH, Johnson EA (2007). "RAD marker microarrays enable rapid mapping of zebrafish mutations". Genome Biology. 8 (6): R105. doi:10.1186/gb-2007-8-6-r105. PMC 2394753. PMID 17553171.
- ^ a b Lewis ZA, Shiver AL, Stiffler N, Miller MR, Johnson EA, Selker EU (October 2007). "High-density detection of restriction-site-associated DNA markers for rapid mapping of mutated loci in Neurospora". Genetics. 177 (2): 1163–1171. doi:10.1534/genetics.107.078147. PMC 2034621. PMID 17660537.
- ^ a b Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA (February 2010). "Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags". PLOS Genetics. 6 (2): e1000862. doi:10.1371/journal.pgen.1000862. PMC 2829049. PMID 20195501.
- ^ a b Shendure J, Ji H (October 2008). "Next-generation DNA sequencing". Nature Biotechnology. 26 (10): 1135–1145. doi:10.1038/nbt1486. PMID 18846087. S2CID 6384349.
- ^ Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012). "Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species". PLOS ONE. 7 (5): e37135. Bibcode:2012PLoSO...737135P. doi:10.1371/journal.pone.0037135. PMC 3365034. PMID 22675423.
- ^ a b Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012). "Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species". PLOS ONE. 7 (5): e37135. Bibcode:2012PLoSO...737135P. doi:10.1371/journal.pone.0037135. PMC 3365034. PMID 22675423.
- ^ Suchan T, Pitteloud C, Gerasimova NS, Kostikova A, Schmid S, Arrigo N, et al. (2016). Orlando L (ed.). "Hybridization Capture Using RAD Probes (hyRAD), a New Tool for Performing Genomic Analyses on Collection Specimens". PLOS ONE. 11 (3): e0151651. Bibcode:2016PLoSO..1151651S. doi:10.1371/journal.pone.0151651. PMC 4801390. PMID 26999359.
- ^ Crates R, Olah G, Adamski M, Aitken N, Banks S, Ingwersen D, et al. (2019). Russello MA (ed.). "Genomic impact of severe population decline in a nomadic songbird". PLOS ONE. 14 (10): e0223953. Bibcode:2019PLoSO..1423953C. doi:10.1371/journal.pone.0223953. PMC 6812763. PMID 31647830.
- ^ Faircloth, Brant C. (2017). Gilbert, M. (ed.). "Identifying conserved genomic elements and designing universal bait sets to enrich them". Methods in Ecology and Evolution. 8 (9): 1103–1112. doi:10.1111/2041-210X.12754. ISSN 2041-210X. S2CID 51896564.
- ^ Olah G, Aitken N, Suchan T (2018-01-25). "hyRAD for birds v1". Protocols.io. doi:10.17504/protocols.io.mt2c6qe.